Hipertensión arterial pulmonar: clasificación de los pacientes
La hipertensión arterial pulmonar (HAP) es una entidad nosológica que tanto cardiólogos, como neumólogos afrontan en la práctica cotidiana dentro del gran grupo de pacientes con hipertensión pulmonar (HP). Su diagnóstico y tratamiento debe ser multidisciplinario, basado en la evidencia actual e individualizado.1,2
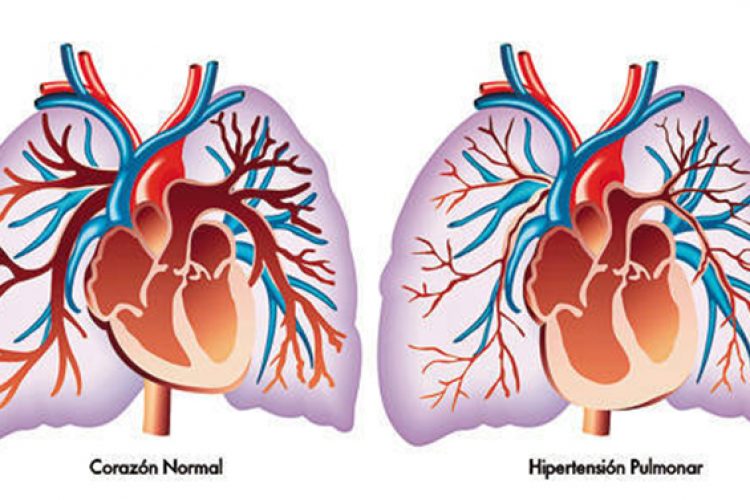
Aunque el diagnóstico de la HAP es por exclusión, es necesario incluir en la definición de los casos los valores de ciertas variables hemodinámicas obtenidas mediante cateterismo cardiaco derecho (CCTD): una presión media de la
arteria pulmonar ≥ 25 mmHg, una presión de oclusión de la arteria pulmonar (POAP) ≤ 15 mmHg y un gasto cardiaco (GC) normal o disminuido.3,4
Durante el Primer Simposium Internacional de la OMS sobre HP en 1973, se clasificó en dos categorías: 1) hipertensión pulmonar primaria (HPP) o 2) HP secundaria, dependiendo de la ausencia o presencia de causas identificables o factores de riesgo. A partir de 1998 (2º Simposio Mundial) se catalogó a la HP en 5 grupos con base en los hallazgos patológicos, características hemodinámicas y similitud en el manejo.
Hubo algunos cambios en el grupo de la Hipertensión Arterial Pulmonar. En el caso de las anemias hemolíticas crónicas: la más importante, la anemia de células falciformes dejaron el grupo 1 para ubicarse en el grupo 5. Además, se actualizó la lista en HAP inducida por fármacos y toxinas. Finalmente se modificó la clasificación de la HAP asociada (HAPA) a cardiopatías congénitas.1
Existen datos de investigación epidemiológica de varios países.5 El estudio ecocardiográfico Armadale (10,314 casos) detectó 936 pacientes con HP (presión sistólica de la arteria pulmonar o PSAP > 40 mmHg), 68% de los casos relacionados con una enfermedad del corazón izquierdo (636 pacientes) y 9% con enfermedad respiratoria (87 pacientes). En los resultados de este estudio se encontró que 2.7% cumplió los criterios clínicos y hemodinámicos de HAP (15 casos/100,000 habitantes) y 0.9% de los casos se catalogó como hipertensión arterial pulmonar idiopática (4.8 casos/100,000 habitantes).6
Referencias
1. Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated clinical classification
of pulmonary hypertension. J Am Coll Cardiol (2013); 62(25 Supl):D34-D41.
2. Howard LS, Graspa J, Dawson D, Bellamy M, Chambers JB, Masani ND, et al. Echocardiographic assessment
of pulmonary hypertension: standard operating procedure. Eur Respir Rev (2012); 21(125):239-248.
3. Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, et al. Definitions and diagnosis of
pulmonary hypertension. J Am Coll Cardiol (2013); 62(25):D42-D50.
4. Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. Guidelines for the diagnosis and
treatment of pulmonary hypertension. Eur Heart J (2009); 30:2493-2537.
5. Mc Goon M, Benza RL, Escribano-Subias P, Jiang X, Miller DP, Peacock AJ, Pepke-Zaba J, et al.Pulmonary
Arterial Hypertension: Epidemiology and Registries. J Am Coll Cardiol (2013); 62(25):D51-59.
6. Strange G, Playford D, Stewart S, Deague JA, Nelson H, Kent A, et al. Pulmonary hypertension: prevalence
and mortality in the Armadale echocardiography cohort. Heart (2012); 98(24):1805-1811.